





Ademais da tecnoloxía, a síntese de glicósidos sempre foi de interese para a ciencia, xa que é unha reacción moi común na natureza. Artigos recentes de Schmidt, Toshima e Tatsuta, así como moitas referencias citadas neles, comentaron unha ampla gama de potenciais sintéticos.
Na síntese de glicósidos, os compoñentes multiazucrados combínanse con nucleófilos, como alcohois, carbohidratos ou proteínas. Se se require unha reacción selectiva cun dos grupos hidroxilo dun carbohidrato, todas as demais funcións deben protexerse no primeiro paso. En principio, os procesos encimáticos ou microbianos, debido á súa selectividade, poden substituír os complexos pasos de protección e desprotección química para separar selectivamente os glicósidos en certas rexións. Non obstante, debido á longa historia dos alquilglicósidos, a aplicación de encimas na síntese de glicósidos non foi amplamente estudada nin aplicada.
Debido á capacidade dos sistemas encimáticos axeitados e aos altos custos de produción, a síntese encimática de alquilpoliglicósidos non está lista para ser actualizada ao nivel industrial, e prefírense os métodos químicos.
En 1870, MaColley informou da síntese de "acetoclorhidrosa" (1, figura 2) por reacción da dextrosa (glicosa) con cloruro de acetilo, o que finalmente levou á historia das rutas de síntese de glicósidos.
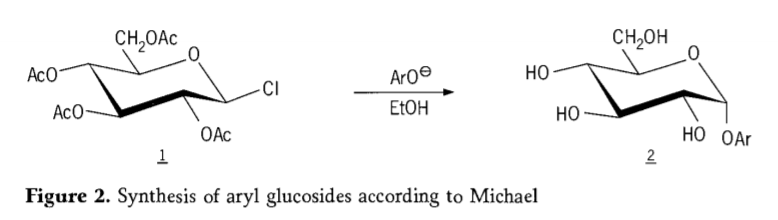
Posteriormente, descubríuse que os haluros de tetra-O-acetil-glucopiranosilo (acetohaloglucosas) eran intermediarios útiles para a síntese estereoselectiva de glucósidos de alquilo puros. En 1879, Arthur Michael conseguiu preparar glicósidos de arilo cristalizables definidos a partir de intermediarios de Colley e fenolatos. (Aro-, Figura 2).
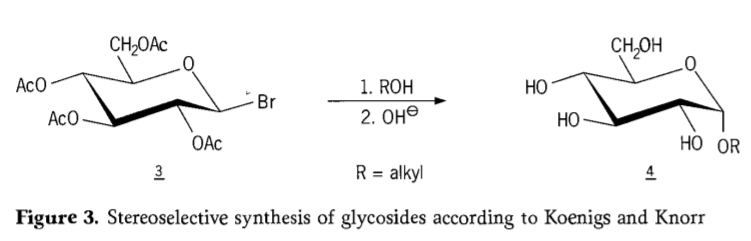
En 1901, a síntese de Michael para unha ampla gama de carbohidratos e aglicóns hidroxílicos, cando W. Koenigs e E. Knorr presentaron o seu proceso de glicosilación estereoselectiva mellorado (Figura 3). A reacción implica unha substitución SN2 no carbono anomérico e prodúcese estereoselectivamente con inversión da configuración, producindo por exemplo o α-glicósido 4 a partir do β-anómero do intermediario aceobromoglicosa 3. A síntese de Koenigs-Knorr ten lugar en presenza de promotores de prata ou mercurio.
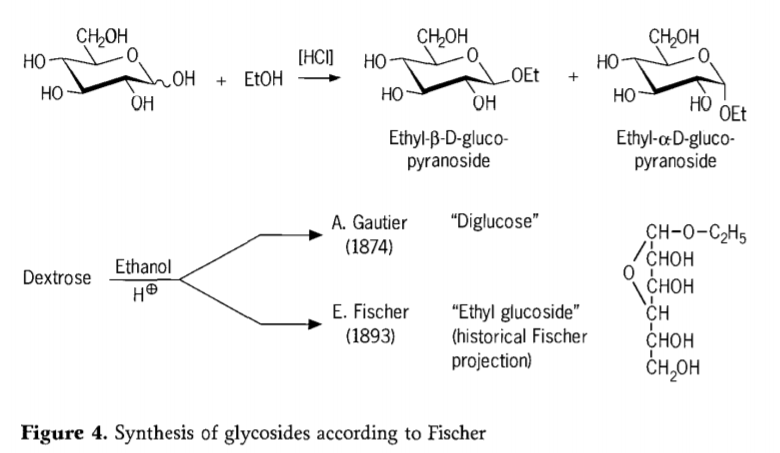
En 1893, Emil Fischer propuxo unha estratexia fundamentalmente diferente para a síntese de alquilglicósidos. Este proceso coñécese hoxe en día como a «glicosilación de Fischer» e comprende unha reacción catalizada por ácidos de glicosas con alcohois. Non obstante, calquera relato histórico debería incluír tamén o primeiro intento rexistrado de A. Gautier en 1874 de converter a dextrosa con etanol anhidro en presenza de ácido clorhídrico. Debido a unha análise elemental enganosa, Gautier cría que obtivera unha «diglicosa». Fischer demostrou máis tarde que a «diglicosa» de Gautier era, en realidade, principalmente etilglicósido (Figura 4).
Fischer definiu correctamente a estrutura do glucósido de etilo, como se pode ver na fórmula furanósidica histórica proposta. De feito, os produtos de glicosilación de Fischer son complexos, na súa maioría mesturas de equilibrio de anómeros α/β e isómeros de piranósido/furanósido que tamén comprenden oligómeros de glicósidos ligados aleatoriamente.
En consecuencia, as especies moleculares individuais non son fáciles de illar das mesturas de reacción de Fischer, o que supuxo un problema grave no pasado. Despois dalgunhas melloras neste método de síntese, Fischer adoptou posteriormente a síntese de Koenigs-Knorr para as súas investigacións. Usando este proceso, E. Fischer e B. Helferich foron os primeiros en informar da síntese dun glucósido de alquilo de cadea longa que presentaba propiedades surfactantes en 1911.
Xa en 1893, Fischer xa observara correctamente as propiedades esenciais dos glicósidos de alquilo, como a súa alta estabilidade fronte á oxidación e á hidrólise, especialmente en medios fortemente alcalinos. Ambas as dúas características son valiosas para os poliglicósidos de alquilo en aplicacións surfactantes.
A investigación relacionada coa reacción de glicosilación aínda está en curso e nos últimos tempos desenvolvéronse varias rutas interesantes para obter glicósidos. Algúns dos procedementos para a síntese de glicósidos resúmense na Figura 5.
En xeral, os procesos de glicosilación química poden dividirse en procesos que levan a equilibrios oligómeros complexos no intercambio de glicosilos catalizado por ácidos.
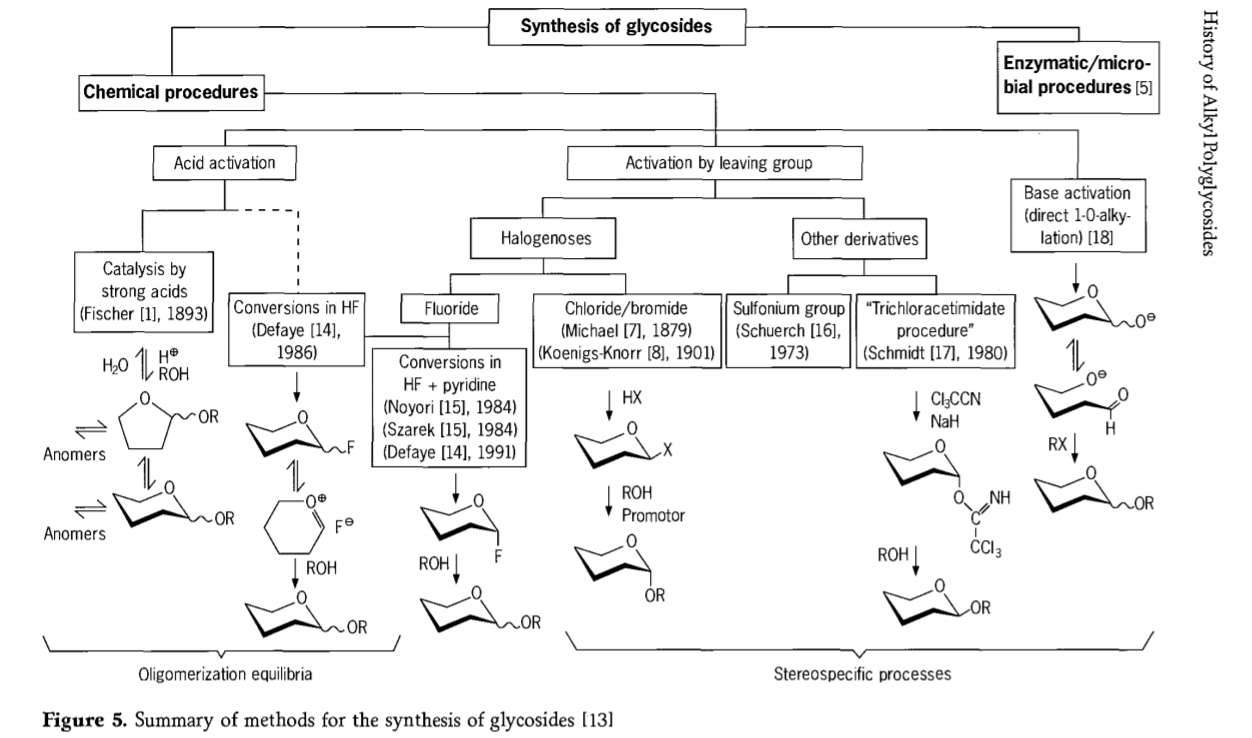
Reaccións en substratos de carbohidratos activados axeitadamente (reaccións glicosídicas de Fischer e reaccións de fluoruro de hidróxeno (HF) con moléculas de carbohidratos desprotexidas) e reaccións de substitución cinéticamente controladas, irreversibles e principalmente estereotáxicas. Un segundo tipo de procedemento pode levar á formación de especies individuais en lugar de mesturas complexas de reaccións, especialmente cando se combina con técnicas de grupos de conservación. Os carbohidratos poden deixar grupos no carbono ectópico, como átomos de halóxeno, sulfonilos ou grupos tricloroacetimidato, ou ser activados por bases antes da conversión en ésteres de triflato.
No caso particular das glicosidacións en fluoruro de hidróxeno ou en mesturas de fluoruro de hidróxeno e piridina (polipiridinio [fluoruro de hidróxeno]), os fluoruros de glicosilo fórmanse in situ e convértense suavemente en glicósidos, por exemplo con alcohois. Demostrouse que o fluoruro de hidróxeno é un medio de reacción fortemente activador e non degradante; obsérvase unha autocondensación de equilibrio (oligomerización) de xeito similar ao proceso de Fischer, aínda que o mecanismo de reacción é probablemente diferente.
Os glicósidos de alquilo quimicamente puros só son axeitados para aplicacións moi especiais. Por exemplo, os glicósidos de alquilo empregáronse con éxito na investigación bioquímica para a cristalización de proteínas de membrana, como a cristalización tridimensional de porina e bacteriorrodopsina en presenza de octil β-D-glucopiranósido (outros experimentos baseados neste traballo levaron a Deisenhofer, Huber e Michel ao Premio Nobel de Química en 1988).
Durante o desenvolvemento dos poliglicósidos de alquilo, empregáronse métodos estereoselectivos a escala de laboratorio para sintetizar unha variedade de substancias modelo e estudar as súas propiedades fisicoquímicas. Debido á súa complexidade, á inestabilidade dos produtos intermedios e á cantidade e natureza crítica dos residuos do proceso, as sínteses do tipo Koenigs-Knorr e outras técnicas de grupos protectores crearían problemas técnicos e económicos significativos. Os procesos de tipo Fischer son comparativamente menos complicados e máis fáciles de levar a cabo a escala comercial e, en consecuencia, son o método preferido para a produción de poliglicósidos de alquilo a grande escala.
Data de publicación: 12 de setembro de 2020